GFP-TRAP based Affinity Chromatography Mass Spectrometry of Drosophila embryos
All entities in our world are connected (just think of the classic "five degrees of Kevin Bacon" experiment). This is also true of proteins within a cell. In order to more fully understand the complex interaction networks and associated flux through pathways that occur during mitotic spindle formation and mitosis, we have developed a simple, yet effective protocol and analysis based on the isolation of GFP-tagged proteins and their interactors, expressed in the developing embryo.
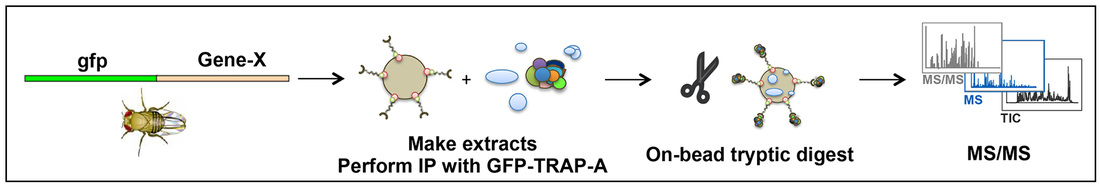
Essentially, embryos expressing the tagged gene of interest are collected, dechorionated, weighed, flash frozen in liquid nitrogen and stored at -80oC, until approximately 0.4g has been collected. The embryos are then homogenised, clarified using high speed (100,000g) centrifugation and the supernatant incubated with 30ul of GFP-TRAP-A beads (Chromotek), equilibrated into buffer, for 2 hrs at 4oC. Following washing, these beads are subjected to Trypsin-based cleavage and processed for Mass Spectrometry, to produce a fingerprint of peptides associated with the beads.
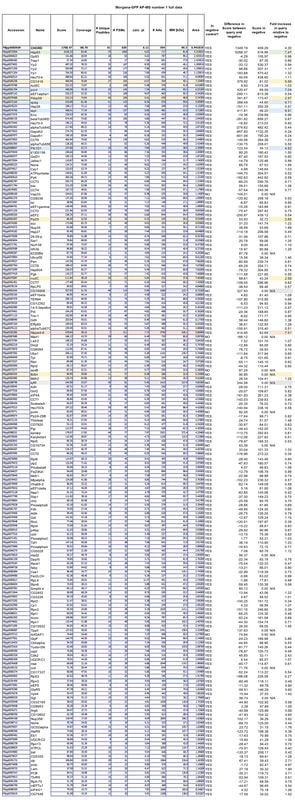
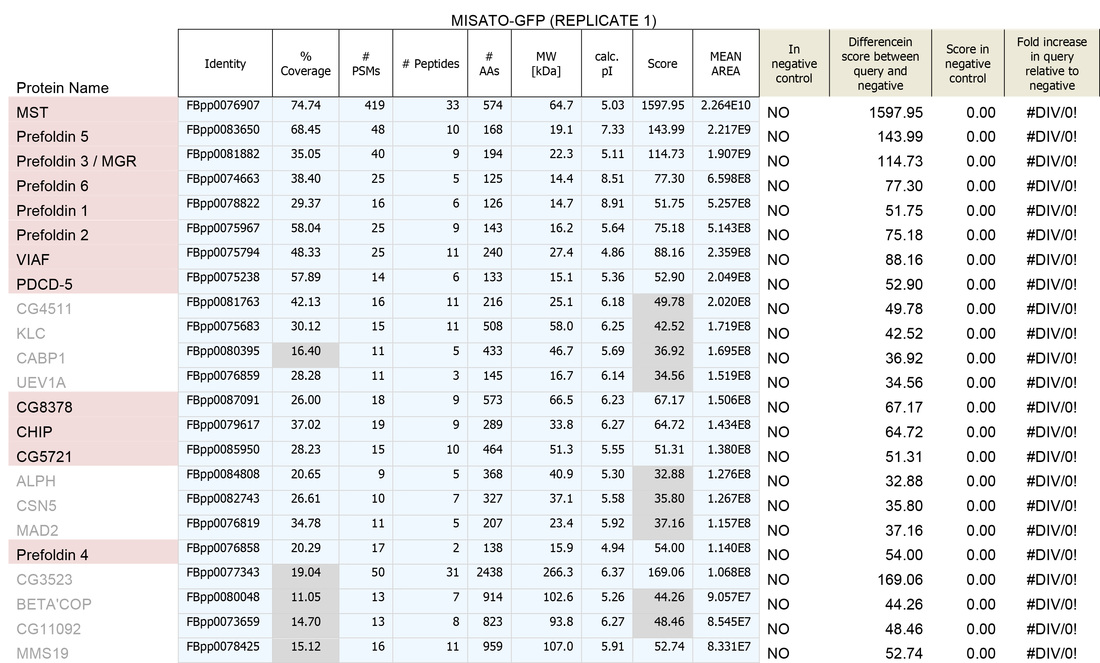
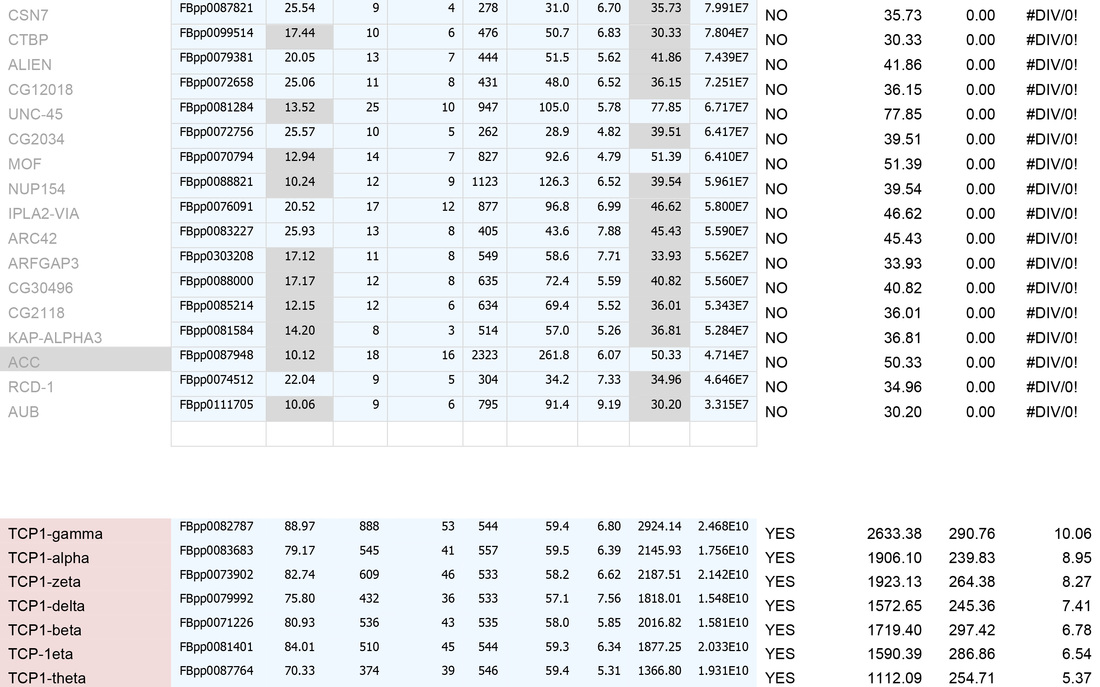
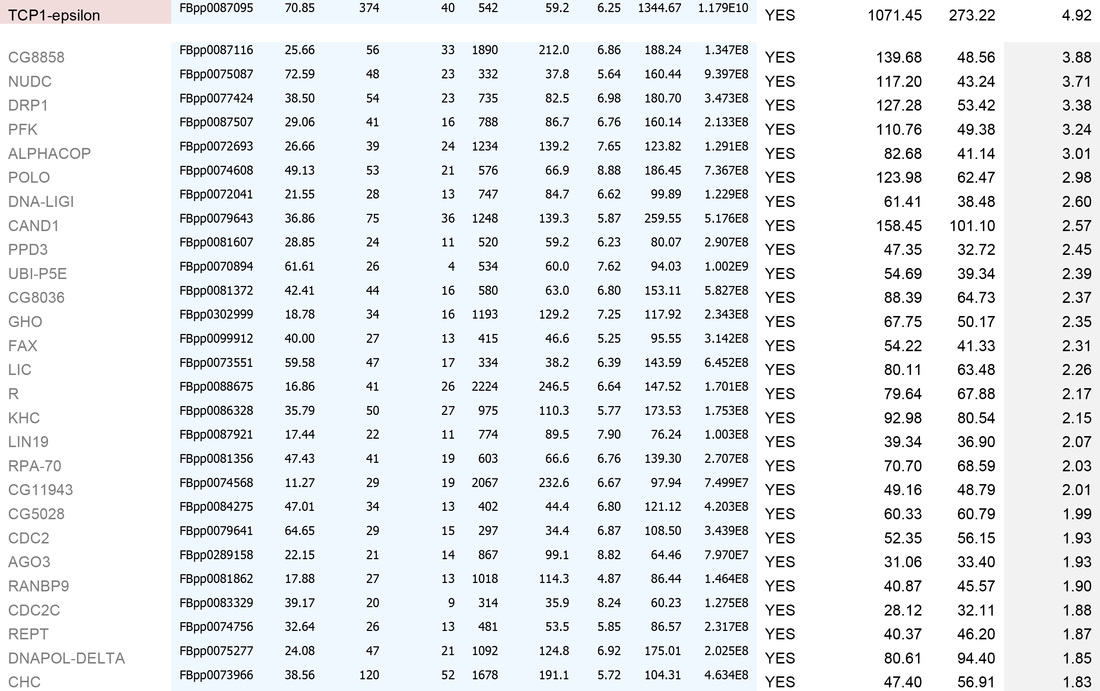
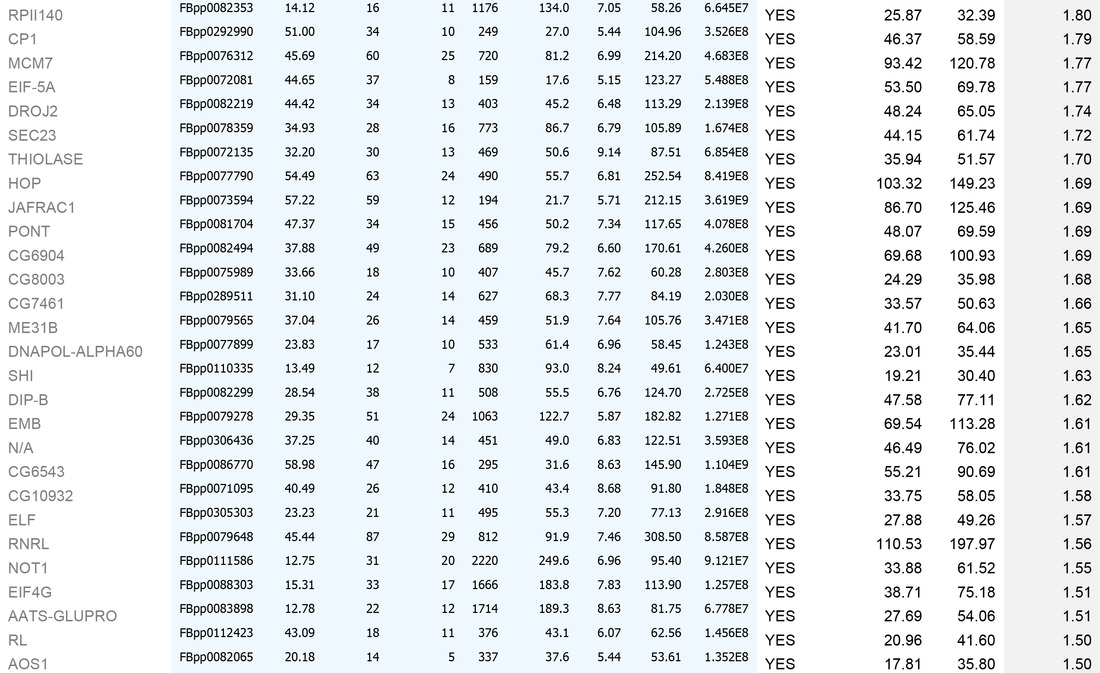
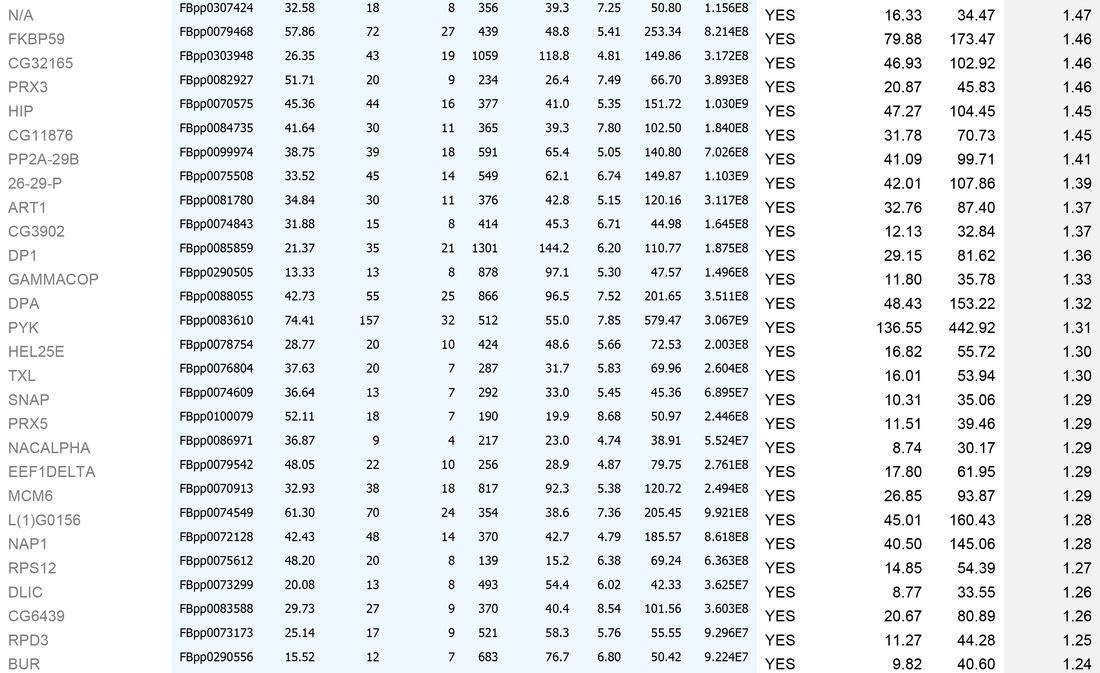
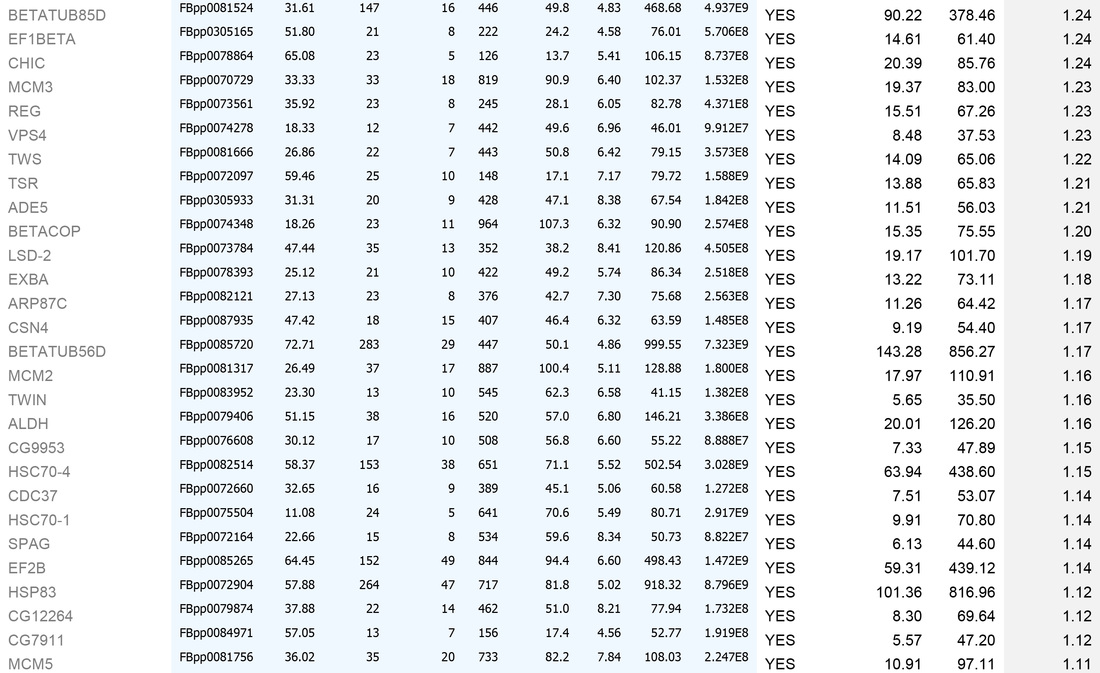
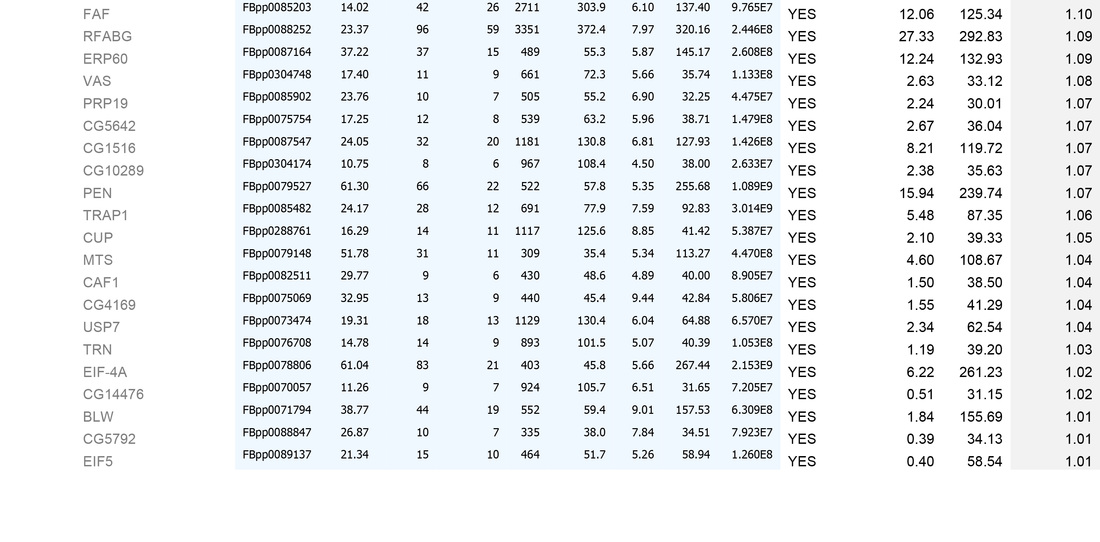
We have currently undertaken this procedure for >30 GFP-tagged fly lines, including for 8 control fly lines. Based on these controls, we have produced a quantitative database of "false positives" - non-specific proteins that are present on GFP-TRAP-A bead precipitates that have been incubated with GFP-transgenic embryo extracts. By cross-referencing individual "bait" protein experiments with this database, we are able to effectively filter out these contaminating proteins, to generate a list of proteins that specifically interact with our protein of interest in the Drosophila embryo (see Table below for a detailed description of how our filtering protocol works, exemplified for an experiment carried out on embryos expressing the protein Misato).
Although this provides a "static" interaction network, we are interested in how this network changes dynamically - through the cell cycle, upon inhibition of particular pathways/modules of mitotic MT generation or upon inhibition of mitotic kinases and phosphatases. The ability to treat dechorionated embryos with drugs, such as MG132, which inhibits the 26S proteasome - arresting embryos in a metaphase-like state - or genetically manipulating them through mutational approaches provides this possibility. We are currently undertaking these types of studies or order to figure out how key mitotic proteins are related to Kevin Bacon (and other interesting questions).
Below are examples of AP-MS we have undertaken, in work that is either published, or under review (Palumbo et al., 2015, Curr Biol - for Misato; Palumbo et al., 2019, under review - for Morgana)
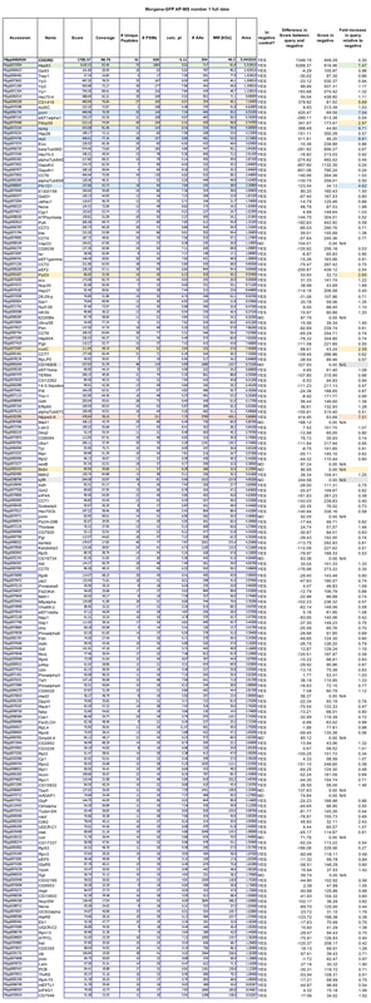
An example dataset after bioinformatics-based filtering:
A. To remove identities that may be "mis-called" through low numbers of attributed peptides, or low scores:
1. Remove any protein with a score of below 30
2. Remove any protein with a single peptide
B. Cross reference the remaining identities against the false-positive database (a list of proteins identified in our negative control experiments)
C. Sort on the basis of whether the identities are present in the false-positive list (YES/NO)
D. For those identities not present in controls, sort by MEAN AREA (the Top 3 Protein Quantification; i.e. the mean of the three highest abundance peptides identified for each protein)
E. For identities that are present in controls, sort by FOLD INCREASE IN QUERY. The higher the value, the more over-represented the identities are in the experimental sample. Remove identities that have <2 fold increase in the experimental sample.
This generates the dataset seen in the Table below.
For high-quality datasets (a judgement call, usually based on high mean area of bait protein), we apply the following additional high stringency filtering:
1. Remove identities that have <20% coverage and scores of <50 (shown in grey below)
2. Remove identities that have <4 fold increase in the experimental sample (shown in grey below)
We then usually focus on identities that have a mean area of within 1000-fold that of the bait protein (i.e. for a bait of 2x1010, concentrate on proteins that have mean areas of 2x108 and above). N.B. This assumes that "important" biological interactions within the cell occur between a minimum of 1000 molecule of bait protein and 1 molecule of interactor.
Essentially, embryos expressing the tagged gene of interest are collected, dechorionated, weighed, flash frozen in liquid nitrogen and stored at -80oC, until approximately 0.4g has been collected. The embryos are then homogenised, clarified using high speed (100,000g) centrifugation and the supernatant incubated with 30ul of GFP-TRAP-A beads (Chromotek), equilibrated into buffer, for 2 hrs at 4oC. Following washing, these beads are subjected to Trypsin-based cleavage and processed for Mass Spectrometry, to produce a fingerprint of peptides associated with the beads.
We have currently undertaken this procedure for >30 GFP-tagged fly lines, including for 8 control fly lines. Based on these controls, we have produced a quantitative database of "false positives" - non-specific proteins that are present on GFP-TRAP-A bead precipitates that have been incubated with GFP-transgenic embryo extracts. By cross-referencing individual "bait" protein experiments with this database, we are able to effectively filter out these contaminating proteins, to generate a list of proteins that specifically interact with our protein of interest in the Drosophila embryo (see Table below for a detailed description of how our filtering protocol works, exemplified for an experiment carried out on embryos expressing the protein Misato).
Although this provides a "static" interaction network, we are interested in how this network changes dynamically - through the cell cycle, upon inhibition of particular pathways/modules of mitotic MT generation or upon inhibition of mitotic kinases and phosphatases. The ability to treat dechorionated embryos with drugs, such as MG132, which inhibits the 26S proteasome - arresting embryos in a metaphase-like state - or genetically manipulating them through mutational approaches provides this possibility. We are currently undertaking these types of studies or order to figure out how key mitotic proteins are related to Kevin Bacon (and other interesting questions).
Below are examples of AP-MS we have undertaken, in work that is either published, or under review (Palumbo et al., 2015, Curr Biol - for Misato; Palumbo et al., 2019, under review - for Morgana)
An example dataset after bioinformatics-based filtering:
A. To remove identities that may be "mis-called" through low numbers of attributed peptides, or low scores:
1. Remove any protein with a score of below 30
2. Remove any protein with a single peptide
B. Cross reference the remaining identities against the false-positive database (a list of proteins identified in our negative control experiments)
C. Sort on the basis of whether the identities are present in the false-positive list (YES/NO)
D. For those identities not present in controls, sort by MEAN AREA (the Top 3 Protein Quantification; i.e. the mean of the three highest abundance peptides identified for each protein)
E. For identities that are present in controls, sort by FOLD INCREASE IN QUERY. The higher the value, the more over-represented the identities are in the experimental sample. Remove identities that have <2 fold increase in the experimental sample.
This generates the dataset seen in the Table below.
For high-quality datasets (a judgement call, usually based on high mean area of bait protein), we apply the following additional high stringency filtering:
1. Remove identities that have <20% coverage and scores of <50 (shown in grey below)
2. Remove identities that have <4 fold increase in the experimental sample (shown in grey below)
We then usually focus on identities that have a mean area of within 1000-fold that of the bait protein (i.e. for a bait of 2x1010, concentrate on proteins that have mean areas of 2x108 and above). N.B. This assumes that "important" biological interactions within the cell occur between a minimum of 1000 molecule of bait protein and 1 molecule of interactor.